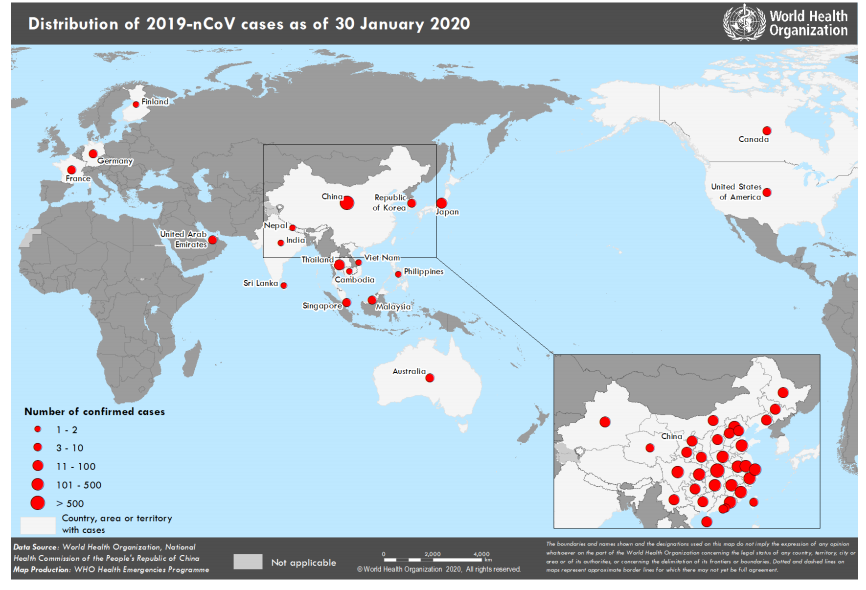
This list is not exhaustive. Mulford librarians are at the ready for consultations to locate information in these resources and information in resources not included below.
Free to all resources
- CDC – Coronavirus – with sections on about the virus, information for healthcare professionals, information for travelers, information for laboratories, information for public health professionals, and news
- The Lancet 2019-nCoV Resource Center – content from Lancet journals as it is published
- BMJ -Coronavirus – includes news, clinical review, infographic, opinion, best practice, analysis, BMJ learning, preprints, and a timeline
- Elsevier Novel Coronavirus Information Center – scientific and medical journals and textbooks, educational products, and a variety of other resources, like travel precautions from the CDC and media posts of interest to our community.
- WHO – Novel Coronavirus (2019-nCoV) with sections Protect Yourself, Travel Advice, Myth-Busters, Situation Reports, and Technical Guidance
Other Resources
- UK Academic Librarian Keith Nockels’ continually updated blog Novel coronavirus (WN-CoV, nCoV) outbreak, China with news, epidemiology and genetics, information for the public, including travel advice, information for health professionals, including travel advice
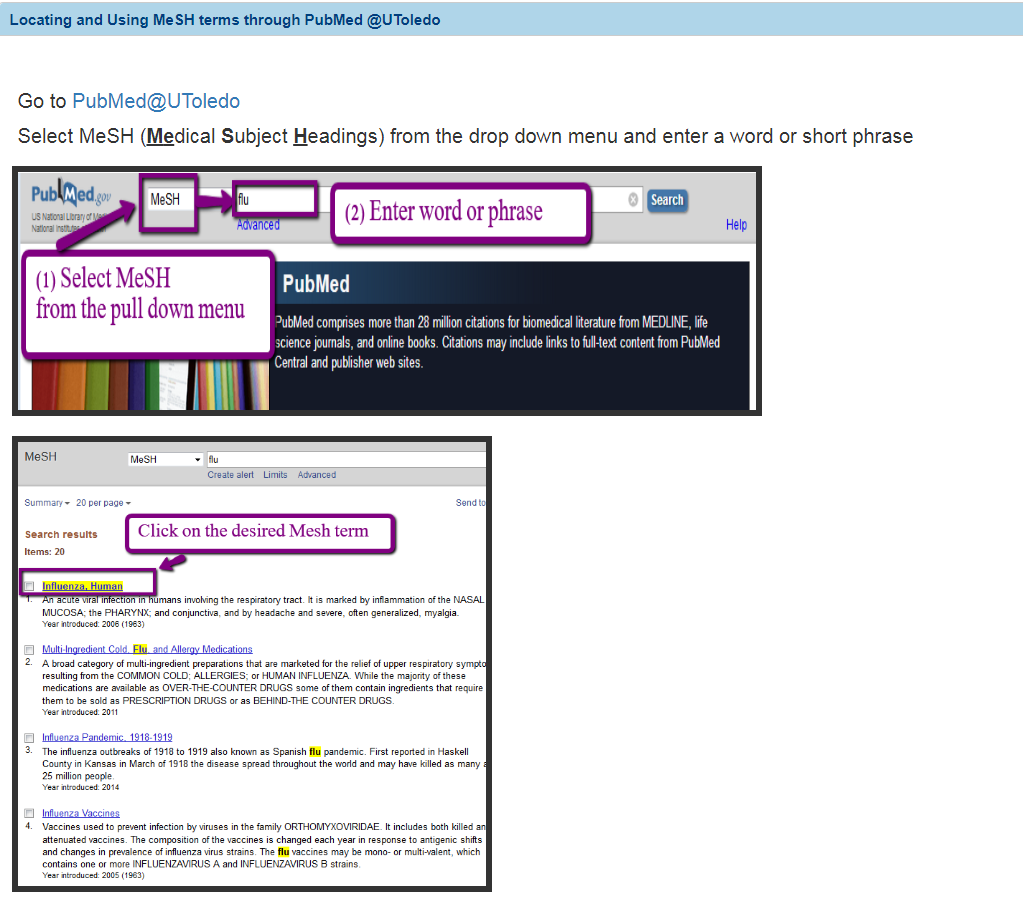
- Searching PubMed@UT
- For articles fully indexed after Feb 13, 2020, enter into the search box…
COVID-19[Supplementary Concept]
(Supplementary concepts may or may not “graduate” to
PubMed’s Medical Subject Headings [MeSH]) - For articles indexed or not indexed before Feb 13, 2020, this may work well
2019-nCoV OR COVID-19 OR (wuhan[tiab] AND coronavirus[tiab])
- For articles fully indexed after Feb 13, 2020, enter into the search box…
Additional and more complete information at the Jan/Feb 2020 National Library of Medicine Technical Bulletin.
Share on Facebook




 The UT Libraries now subscribes to the
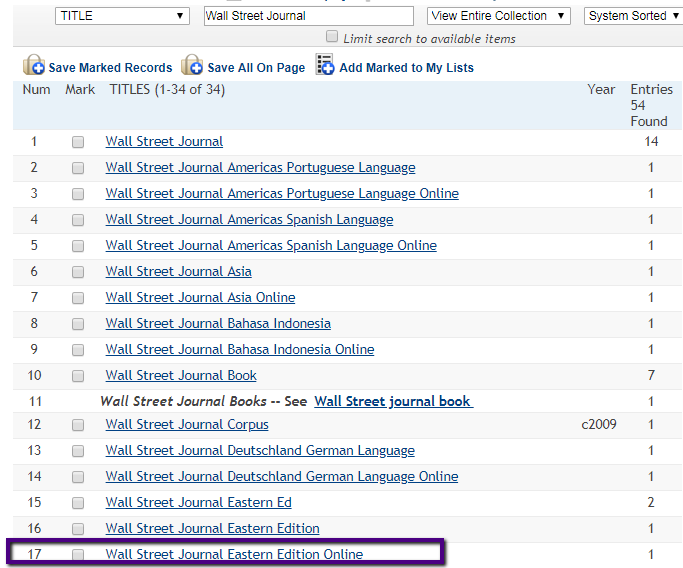
The UT Libraries now subscribes to the  Select Title from the drop down menu and enter Wall Street Journal.
Select Title from the drop down menu and enter Wall Street Journal.